Molecular docking is one of the most commonly used computational technologies in modern drug discovery, it aims to assess interactions between small-molecule ligands and macromolecular target proteins. There are three main types of molecular docking, namely rigid docking, semi-flexible docking and flexible docking. The majority of current molecular docking tools and programs adopt semi-flexible docking schemes, the same goes for Autodock-koto.
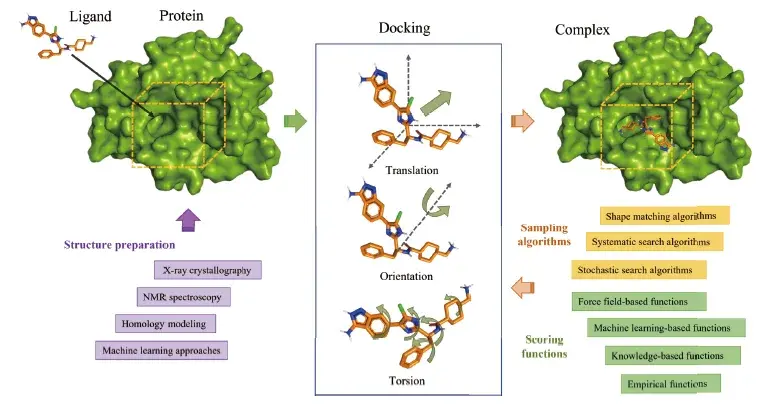
The docking progress can be described as follow: First, a receptor protein and a small molecule ligand are provided with their spatial information, the position of the active site of the receptor also needs to be determined. Then, the structures of complex conformations are determined by the ligand’s translation, orientations, and torsions. The former two correspondingly describe the position and rotation of the ligand as a rigid body, and the latter measures the rotatable bonds between the fragments in the ligand. Finally, the translation, rotation, and intramolecular rotatable bonds of the ligand are adjusted by the search algorithm to pursue the lowest energy among a large number of candidate conformations, according to scoring functions.
Essentially, the molecular docking process can be regarded as an optimization problem, where the search complexity grows exponentially with the increasing degrees of freedom of the ligands. Also, molecular docking is a typical non-deterministic polynomial-time (NP)-hard problem. Thus, it is too expensive to explore all the potential conformations. Only approximate solutions can be obtained by computational sampling algorithms.
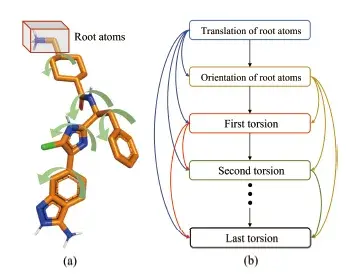
Evolutionary computation (EC) is a very powerful heuristic algorithm that is widely used in engineering control, machine learning, and function optimization. But in the molecular docking problem, the docking performance of EC suffers from the linkage problem and the curse of dimensionality. Here, we proposed a gradient boosting differential evolution approach (Autodock-Koto) for molecular docking, significantly improved docking accuracy and performance of highly flexible ligands.
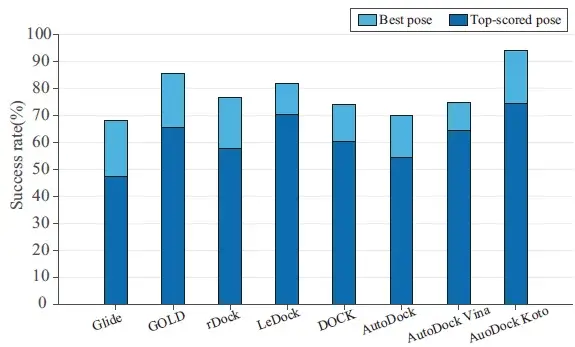
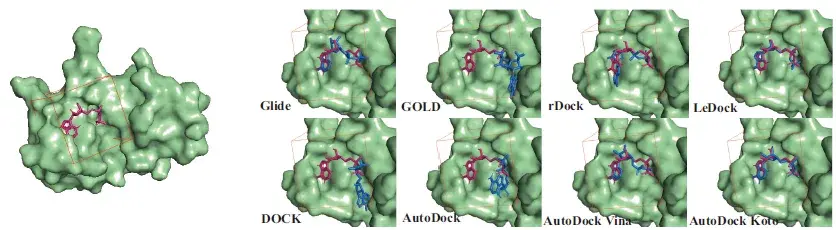
Aotudock-Koto was compared with seven other widely used docking programs and performed the best.
For model details and experimental results, please read the paper “AutoDock Koto: A Gradient Boosting Differential Evolution for Molecular Docking” View paper, the source code can be found at: https://github.com/codezhouj/Molecular_Docking.
Papers
Ji, Junkai, Jin Zhou, Zhangfan Yang, Qiuzhen Lin, Carlos A. Coello Coello. “Autodock koto: A gradient boosting differential evolution for molecular docking.” IEEE Transactions on Evolutionary Computation (2022). View paper
Jin Zhou, Zhangfan Yang, Ying He, Junkai Ji, Qiuzhen Lin, Jianqiang Li. “A novel molecular docking program based on a multi-swarm competitive algorithm.” Swarm and Evolutionary Computation(2023). View paper
Zhangfan Yang, Kun Cao, Junkai Ji, Zexuan Zhu, Jianqiang Li. “Adopting Autodock Koto for Virtual Screening of COVID-19.” International Conference on Intelligent Computing (2023). View paper